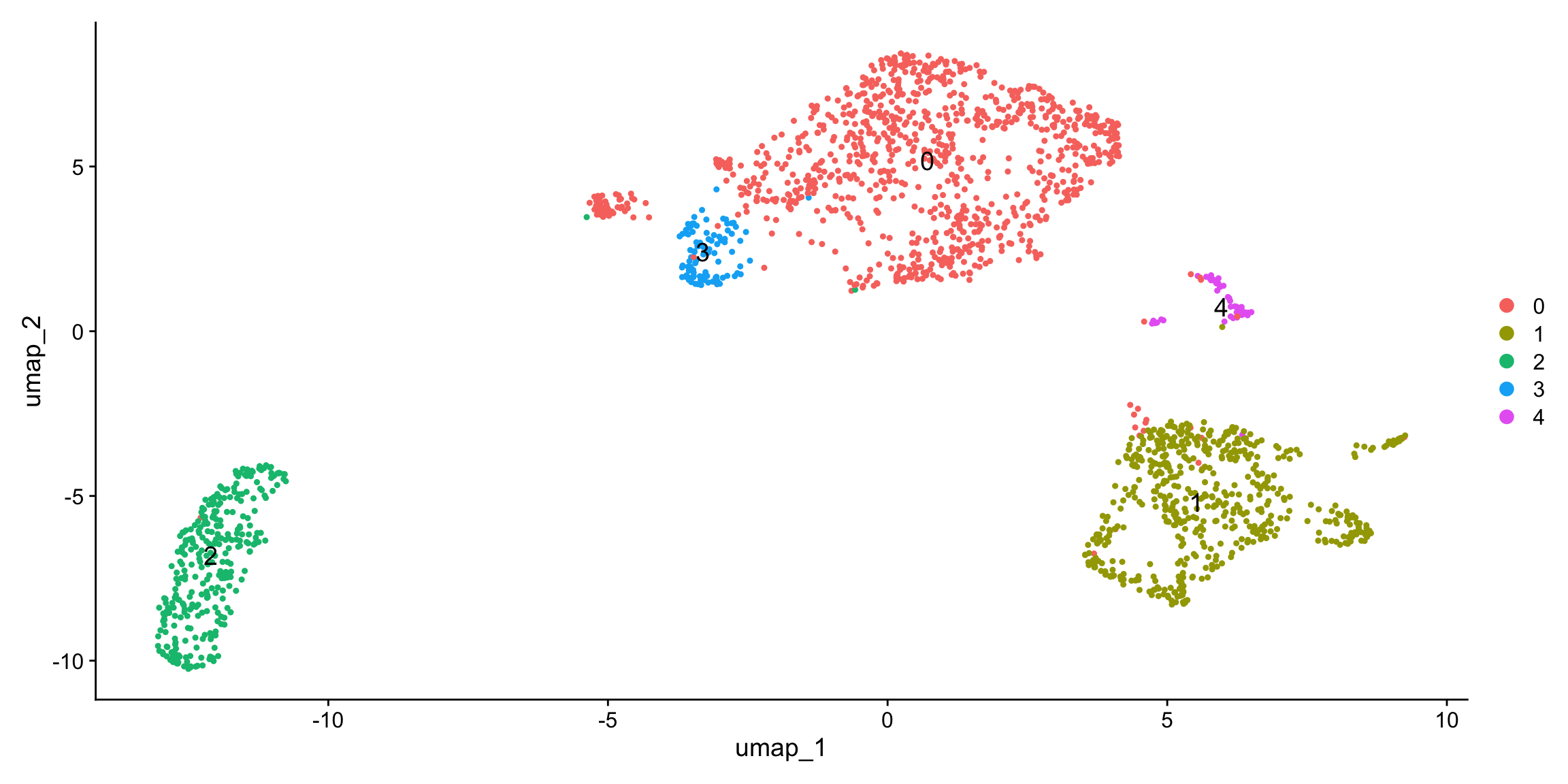
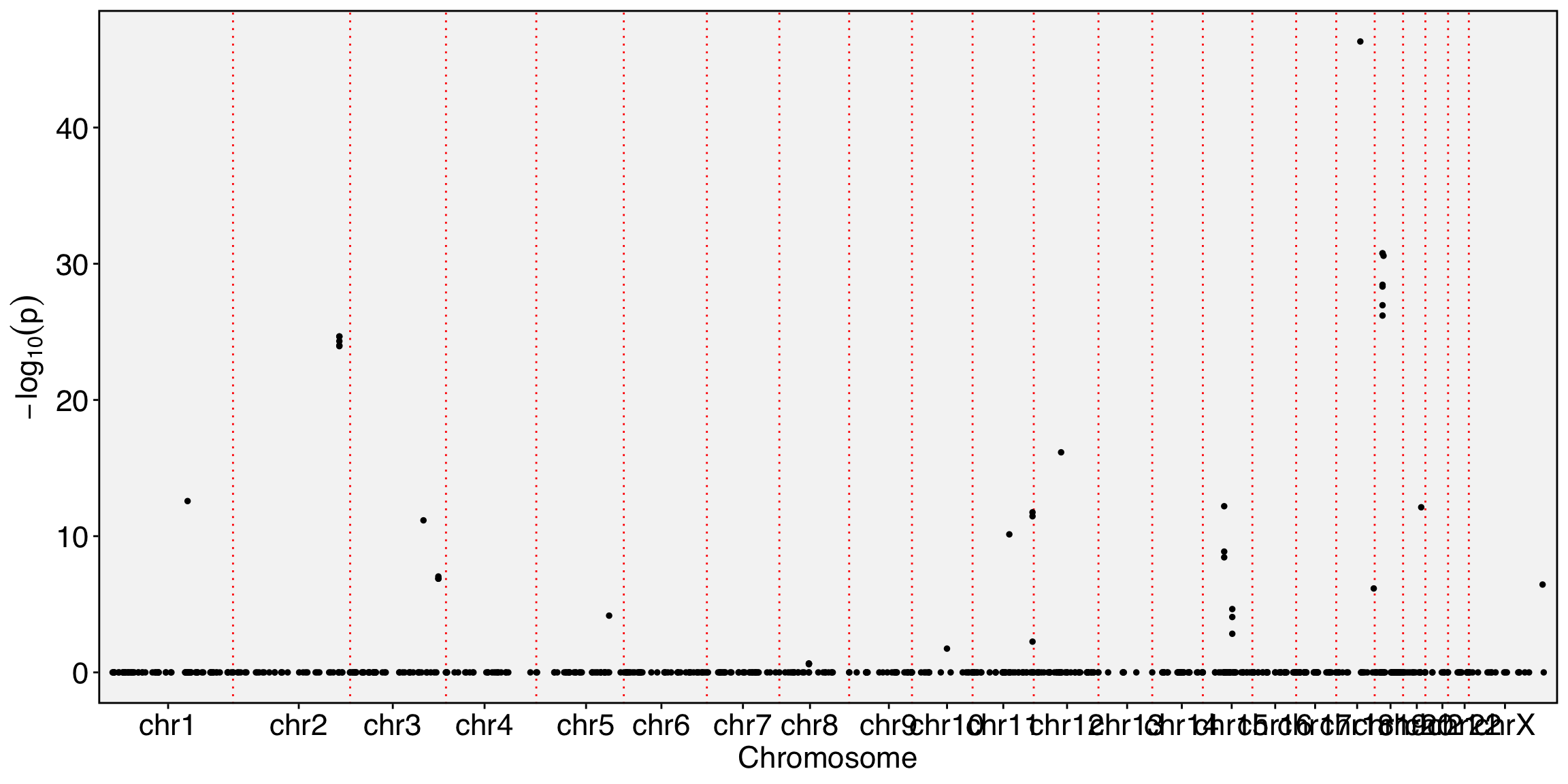
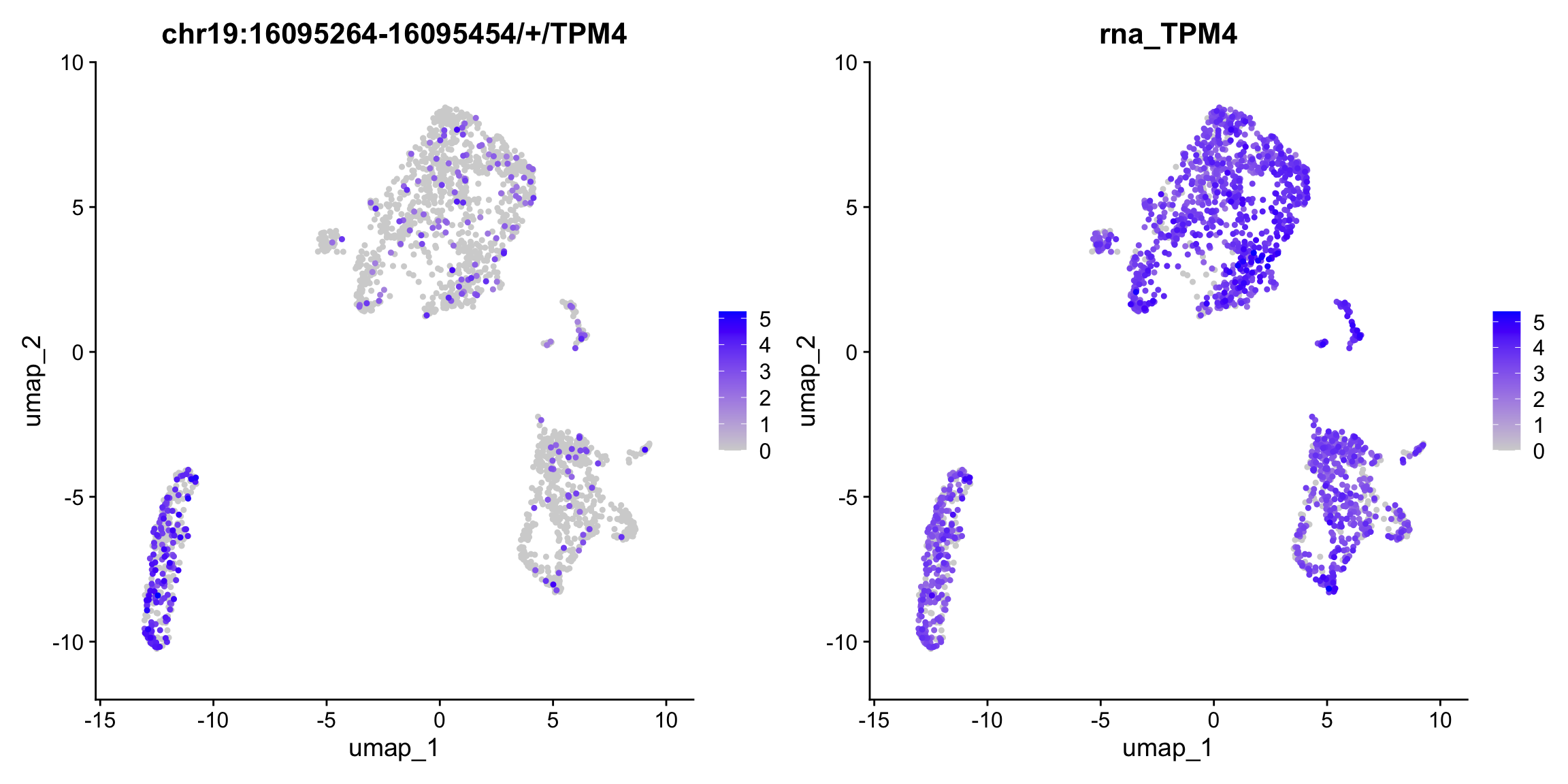
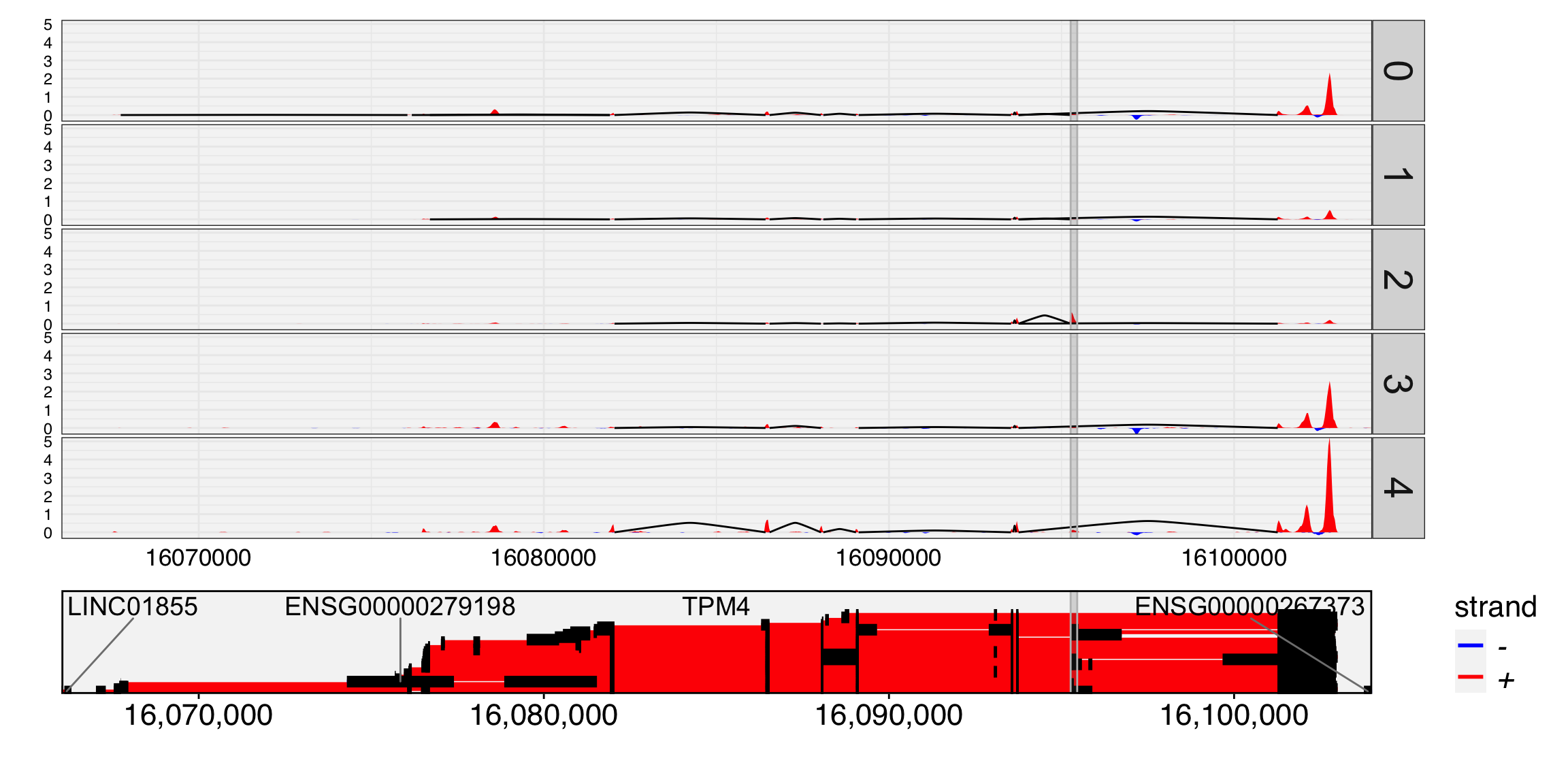
Yano represents an R/C toolkit designed for conducting spatial dissimilarity analysis on single-cell RNA sequencing data. This method revolves around the core concept of examining the distinct expression patterns of a given feature (e.g. exon, snp allele) in relation to its associated binding feature (typically a gene or another allele at the same genomic locus) within the context of cell lineage (1D), spatial position (2D), or the multi-dimensional PCA space. The discernible differences in feature expression patterns and their binding features provide insights into a range of biological phenomena, including alternative splicing, cis-antisense RNA regulation, allele-specific gene expression, and more.
Yano is seamlessly integrated with Seurat, building upon the Seurat object’s framework. Users can perform conventional cell clustering analyses using the state-of-the-art Seurat pipeline and then incorporate exon, SNP counts as new “assays” within the Seurat objects. Subsequently, Yano facilitates the assessment of spatial dissimilarity between these two assays. For more details about the method, please refer to our manuscript.
INSTALL
if (!require("BiocManager")) install.packages('BiocManager') BiocManager::install("shiquan/Yano")
Rewrite TrackPlot() highlight rendering with grid primitives (rectGrob + segmentsGrob) for cross-track spanning. Highlights now span continuously across all facet panels and tracks (coverage, BED, gene annotation) with visual protrusion. Multiple overlapping highlights use additive alpha blending with dark orange boundary lines.
CRAN preparation: fix examples, NAMESPACE, DESCRIPTION metadata, and documentation issues. Register native routines (src/init.c) for CRAN compliance.
Add gtable to Imports, systemfonts to Suggests, remove unused R.utils.
New S3 methods: print.TrackPlot and grid.draw.TrackPlot.
Fix combined & theme() assignment bug and data.frame highlights parsing.
Update README with package overview and key capabilities.
Updated vignette for grid-based highlights; rendered all documentation with Quarto.
1.4 2026/06/11
New functions: GetWeightsFromCoords(), GetWeightsFromSNN() for flexible weight matrix construction.
Improve TrackPlot() highlight continuity and y-axis label readability.
Harden weights.R with input validation and boundary checking.
Update LICENSE to Yano authors (2023–2026).
1.3 2026/06/05
Update citation from bioRxiv to Cell Reports Methods.
Simplified parameters for RunCorrBlock() to streamlines the function and makes it more concise. In addition, new selector functions, *Selector(), introduced.