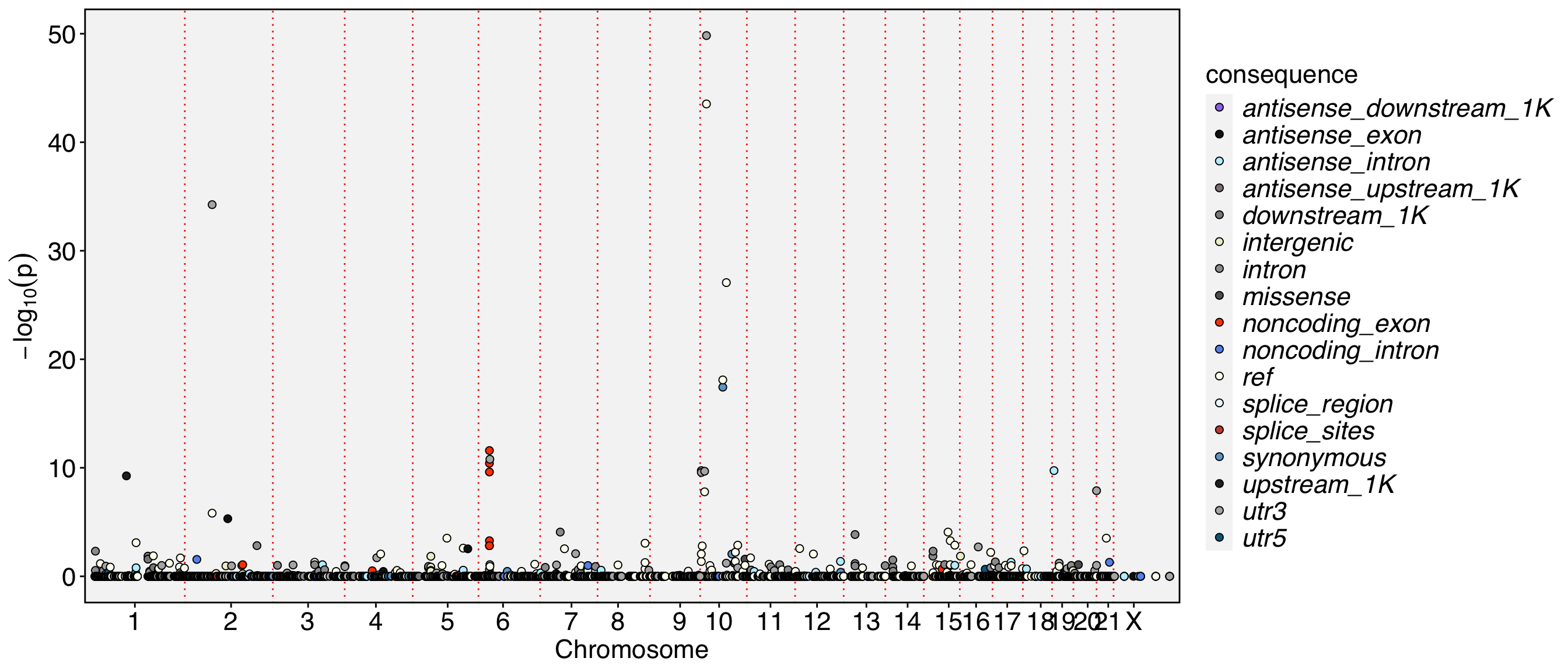
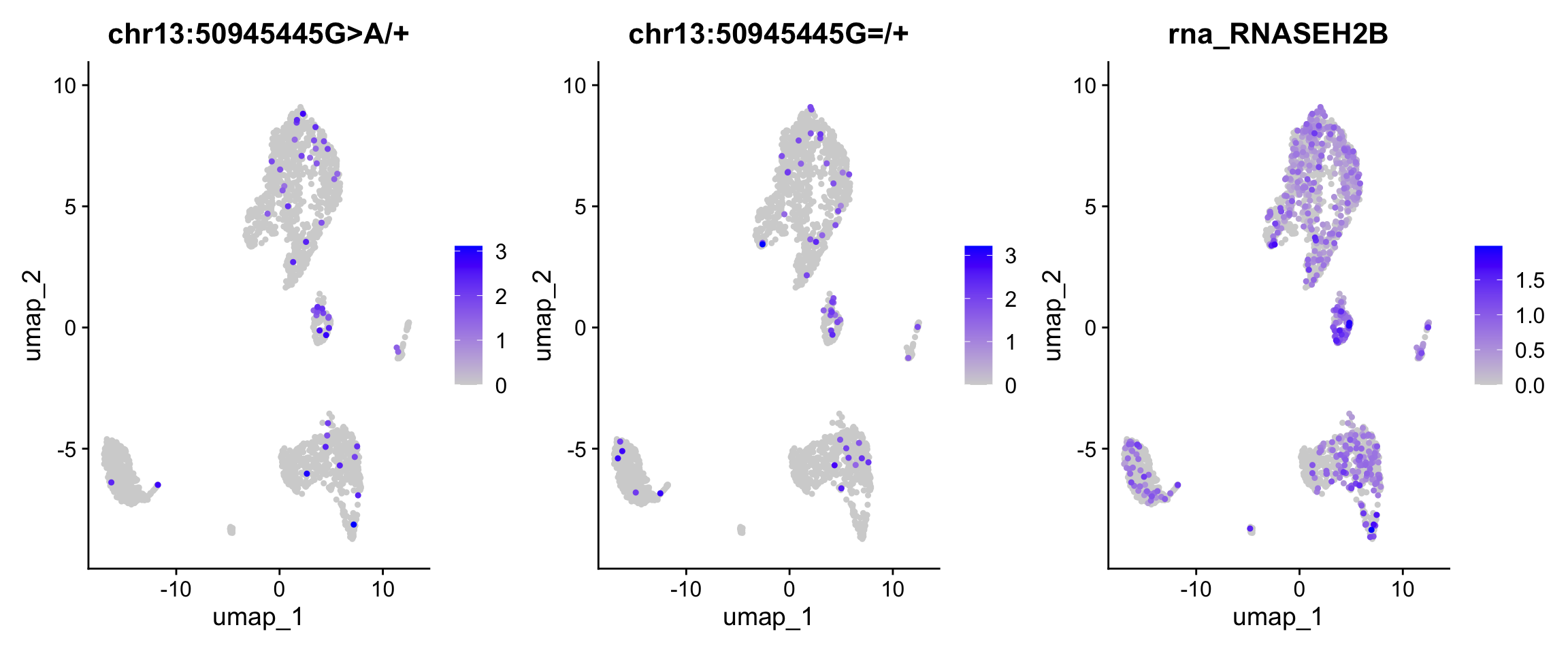
##fileformat=VCFv4.1
##FILTER=<ID=PASS,Description="All filters passed">
##fileDate=2025-02-17
##source=ClinVar
##reference=GRCh38
##ID=<Description="ClinVar Variation ID">
##INFO=<ID=AF_ESP,Number=1,Type=Float,Description="allele frequencies from GO-ESP">
##INFO=<ID=AF_EXAC,Number=1,Type=Float,Description="allele frequencies from ExAC">
##INFO=<ID=AF_TGP,Number=1,Type=Float,Description="allele frequencies from TGP">
##INFO=<ID=ALLELEID,Number=1,Type=Integer,Description="the ClinVar Allele ID">
##INFO=<ID=CLNDN,Number=.,Type=String,Description="ClinVar's preferred disease name for the concept specified by disease identifiers in CLNDISDB">
##INFO=<ID=CLNDNINCL,Number=.,Type=String,Description="For included Variant : ClinVar's preferred disease name for the concept specified by disease identifiers in CLNDISDB">
##INFO=<ID=CLNDISDB,Number=.,Type=String,Description="Tag-value pairs of disease database name and identifier submitted for germline classifications, e.g. OMIM:NNNNNN">
##INFO=<ID=CLNDISDBINCL,Number=.,Type=String,Description="For included Variant: Tag-value pairs of disease database name and identifier for germline classifications, e.g. OMIM:NNNNNN">
##INFO=<ID=CLNHGVS,Number=.,Type=String,Description="Top-level (primary assembly, alt, or patch) HGVS expression.">
##INFO=<ID=CLNREVSTAT,Number=.,Type=String,Description="ClinVar review status of germline classification for the Variation ID">
##INFO=<ID=CLNSIG,Number=.,Type=String,Description="Aggregate germline classification for this single variant; multiple values are separated by a vertical bar">
##INFO=<ID=CLNSIGCONF,Number=.,Type=String,Description="Conflicting germline classification for this single variant; multiple values are separated by a vertical bar">
##INFO=<ID=CLNSIGINCL,Number=.,Type=String,Description="Germline classification for a haplotype or genotype that includes this variant. Reported as pairs of VariationID:classification; multiple values are separated by a vertical bar">
##INFO=<ID=CLNSIGSCV,Number=.,Type=String,Description="SCV accession numbers for the submissions that contribute to the aggregate germline classification in ClinVar; multiple values are separated by a vertical bar">
##INFO=<ID=CLNVC,Number=1,Type=String,Description="Variant type">
##INFO=<ID=CLNVCSO,Number=1,Type=String,Description="Sequence Ontology id for variant type">
##INFO=<ID=CLNVI,Number=.,Type=String,Description="the variant's clinical sources reported as tag-value pairs of database and variant identifier">
##INFO=<ID=DBVARID,Number=.,Type=String,Description="nsv accessions from dbVar for the variant">
##INFO=<ID=GENEINFO,Number=1,Type=String,Description="Gene(s) for the variant reported as gene symbol:gene id. The gene symbol and id are delimited by a colon (:) and each pair is delimited by a vertical bar (|)">
##INFO=<ID=MC,Number=.,Type=String,Description="comma separated list of molecular consequence in the form of Sequence Ontology ID|molecular_consequence">
##INFO=<ID=ONCDN,Number=.,Type=String,Description="ClinVar's preferred disease name for the concept specified by disease identifiers in ONCDISDB">
##INFO=<ID=ONCDNINCL,Number=.,Type=String,Description="For included variant: ClinVar's preferred disease name for the concept specified by disease identifiers in ONCDISDBINCL">
##INFO=<ID=ONCDISDB,Number=.,Type=String,Description="Tag-value pairs of disease database name and identifier submitted for oncogenicity classifications, e.g. MedGen:NNNNNN">
##INFO=<ID=ONCDISDBINCL,Number=.,Type=String,Description="For included variant: Tag-value pairs of disease database name and identifier for oncogenicity classifications, e.g. OMIM:NNNNNN">
##INFO=<ID=ONC,Number=.,Type=String,Description="Aggregate oncogenicity classification for this single variant; multiple values are separated by a vertical bar">
##INFO=<ID=ONCINCL,Number=.,Type=String,Description="Oncogenicity classification for a haplotype or genotype that includes this variant. Reported as pairs of VariationID:classification; multiple values are separated by a vertical bar">
##INFO=<ID=ONCREVSTAT,Number=.,Type=String,Description="ClinVar review status of oncogenicity classification for the Variation ID">
##INFO=<ID=ONCSCV,Number=.,Type=String,Description="SCV accession numbers for the submissions that contribute to the aggregate oncogenicity classification in ClinVar; multiple values are separated by a vertical bar">
##INFO=<ID=ONCCONF,Number=.,Type=String,Description="Conflicting oncogenicity classification for this single variant; multiple values are separated by a vertical bar">
##INFO=<ID=ORIGIN,Number=.,Type=String,Description="Allele origin. One or more of the following values may be added: 0 - unknown; 1 - germline; 2 - somatic; 4 - inherited; 8 - paternal; 16 - maternal; 32 - de-novo; 64 - biparental; 128 - uniparental; 256 - not-tested; 512 - tested-inconclusive; 1073741824 - other">
##INFO=<ID=RS,Number=.,Type=String,Description="dbSNP ID (i.e. rs number)">
##INFO=<ID=SCIDN,Number=.,Type=String,Description="ClinVar's preferred disease name for the concept specified by disease identifiers in SCIDISDB">
##INFO=<ID=SCIDNINCL,Number=.,Type=String,Description="For included variant: ClinVar's preferred disease name for the concept specified by disease identifiers in SCIDISDBINCL">
##INFO=<ID=SCIDISDB,Number=.,Type=String,Description="Tag-value pairs of disease database name and identifier submitted for somatic clinial impact classifications, e.g. MedGen:NNNNNN">
##INFO=<ID=SCIDISDBINCL,Number=.,Type=String,Description="For included variant: Tag-value pairs of disease database name and identifier for somatic clinical impact classifications, e.g. OMIM:NNNNNN">
##INFO=<ID=SCIREVSTAT,Number=.,Type=String,Description="ClinVar review status of somatic clinical impact for the Variation ID">
##INFO=<ID=SCI,Number=.,Type=String,Description="Aggregate somatic clinical impact for this single variant; multiple values are separated by a vertical bar">
##INFO=<ID=SCIINCL,Number=.,Type=String,Description="Somatic clinical impact classification for a haplotype or genotype that includes this variant. Reported as pairs of VariationID:classification; multiple values are separated by a vertical bar">
##INFO=<ID=SCISCV,Number=.,Type=String,Description="SCV accession numbers for the submissions that contribute to the aggregate somatic clinical impact in ClinVar; multiple values are separated by a vertical bar">
##contig=<ID=1>
##contig=<ID=2>
##contig=<ID=3>
##contig=<ID=4>
##contig=<ID=5>
##contig=<ID=6>
##contig=<ID=7>
##contig=<ID=8>
##contig=<ID=9>
##contig=<ID=10>
##contig=<ID=11>
##contig=<ID=12>
##contig=<ID=13>
##contig=<ID=14>
##contig=<ID=15>
##contig=<ID=16>
##contig=<ID=17>
##contig=<ID=18>
##contig=<ID=19>
##contig=<ID=20>
##contig=<ID=21>
##contig=<ID=22>
##contig=<ID=X>
##contig=<ID=Y>
##contig=<ID=MT>
##contig=<ID=NT_113889.1>
##contig=<ID=NT_187633.1>
##contig=<ID=NT_187661.1>
##contig=<ID=NT_187693.1>
##contig=<ID=NW_009646201.1>
##bcftools_viewVersion=1.19+htslib-1.19
##bcftools_viewCommand=view clinvar.vcf.gz 15:92992967-92992967; Date=Mon Jun 15 14:43:34 2026
#CHROM POS ID REF ALT QUAL FILTER INFO
15 92992967 633515 C G . . ALLELEID=621980;CLNDISDB=MedGen:CN517202;CLNDN=not_provided;CLNHGVS=NC_000015.10:g.92992967C>G;CLNREVSTAT=criteria_provided,_single_submitter;CLNSIG=Pathogenic;CLNSIGSCV=SCV000920484;CLNVC=single_nucleotide_variant;CLNVCSO=SO:0001483;CLNVI=ClinGen:CA393896759;GENEINFO=CHD2:1106;MC=SO:0001587|nonsense;ORIGIN=1;RS=2272457
15 92992967 257709 C T . . AF_ESP=0.2485;AF_EXAC=0.22658;AF_TGP=0.19349;ALLELEID=255446;CLNDISDB=MeSH:D030342,MedGen:C0950123|MONDO:MONDO:0014150,MedGen:C3809278,OMIM:615369,Orphanet:1942,Orphanet:2382|MedGen:C3661900|MedGen:CN169374;CLNDN=Inborn_genetic_diseases|Developmental_and_epileptic_encephalopathy_94|not_provided|not_specified;CLNHGVS=NC_000015.10:g.92992967C>T;CLNREVSTAT=criteria_provided,_multiple_submitters,_no_conflicts;CLNSIG=Benign;CLNSIGSCV=SCV000307175|SCV000519223|SCV000841518|SCV000846021|SCV001730597|SCV001806395|SCV005087698|SCV005297510;CLNVC=single_nucleotide_variant;CLNVCSO=SO:0001483;CLNVI=ClinGen:CA7748248;GENEINFO=CHD2:1106;MC=SO:0001819|synonymous_variant;ORIGIN=1;RS=2272457